시퀀싱이 완료된 후 파일을 전달받아서 demux 를 하면 해당 sequencing run 에서의 seqeuncing quality 를 확인할 수 있습니다.
퀄리티를 확인한 뒤 quality control tools 를 사용해서 300 bp 의 foward, reverse read 의 어느 정도를 자르고, 어느 정도를 가지고 분석할것인지를 선택할 수 있습니다. (저의 경우에는 qiime2 에서 DADA2 를 사용합니다.)
이 과정에서 quanlity score 에 대한 것은 illumina 홈페이지에서 확인할 수 있습니다.

sequencing quality score 란, 각각의 염기가 잘못 호출될 가능성을 나타내는 지표로 각각의 염기에 대해 phred-like algorithm 을 사용하여 가능성을 측정합니다.
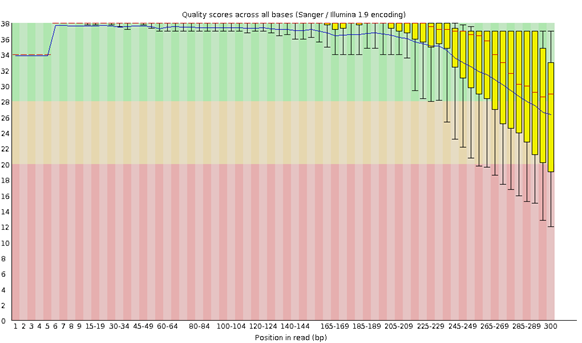
.qzv 파일을 qiime2 view 로 확인하면 다음과 같은 그래프를 확인할 수 있는데 y 축의 quality score 가 phred-like algorithm 으로 계산된 것입니다.
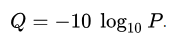
Q (quality score) 와 P (염기의 호출 (base call) 이 잘못될 확률) 의 관계는 위와 같고 조금 더 직관적으로는 아래 표에서의 관계와 같습니다.

Q 가 10 일때, 염기 호출에 대한 정확도는 90%
Q 가 20 일때, 염기 호출에 대한 정확도는 99%
.
.
.
로 나타나며 어느 정도까지 허용할지는 사용자에 따라 다르지만, illumina 에서는 30 정도를 허용가능하다고 보고 있습니다.
이렇게 오류에 대한 가능성이 있는 이유는, flow cell 에서 시퀀싱을 하는 과정에서 나타나는 chemical reactions 이 불완전할 수 있기 때문입니다.
예를 들어, 뉴클레오타이드에 따라 다르게 나타나는 fluorescent signal 을 탐지하는 과정에서 noies, artifacts 등으로 인한 불확실함 등이 있을 수 있습니다.
실제 dada2 를 통하여 퀄리티가 낮은 부분들을 자를 수 있습니다. 아래와 같은 코드로 실행하며, trim 은 5', truncate 는 3' 의 부분을 자른다는 의미입니다.
qiime dada2 denoise-paired
--i-demultiplexed-seqs 'demux_seq'
--p-trim-left-f 5
--p-trim-left-r 5
--p-trunc-len-f 250
--p-trunc-len-r 230
--output-dir "output_dir"
각 reads 는 PCR 에 의해 합성되므로, 5' 부분이 일반적으로 quality score 가 더 높아 조금만 자르게 되고,
경험적으로 forward 보다는 reverse 가 quality score 가 낮습니다.
# reference
1) Ewing, Brent, and Phil Green. "Base-calling of automated sequencer traces using phred. II. Error probabilities." Genome research 8.3 (1998): 186-194.
'bioinfo' 카테고리의 다른 글
| Sequencing quality (Illumina Miseq) : 2 (0) | 2023.09.02 |
|---|---|
| Sequencing quality (Illumina Miseq) : 1 (0) | 2023.09.01 |
| 차원축소 (PCA, PCoA, LDA) in microbiome data 와 LEfSe : 2 (microbiome) (0) | 2023.08.23 |
| 차원축소 (PCA, PCoA, LDA) in microbiome data 와 LEfSe : 1 (microbiome) (0) | 2023.08.21 |
| differential abundance tools for microbiome data (상대적 풍부도) (0) | 2023.08.20 |