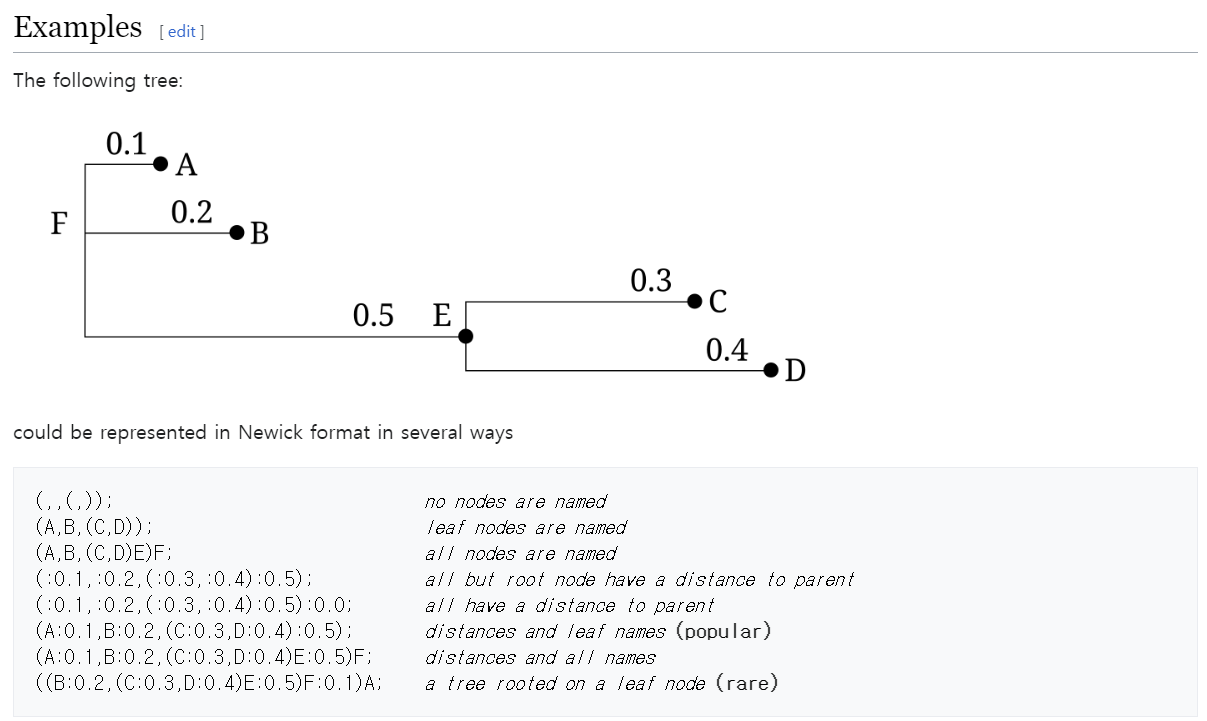
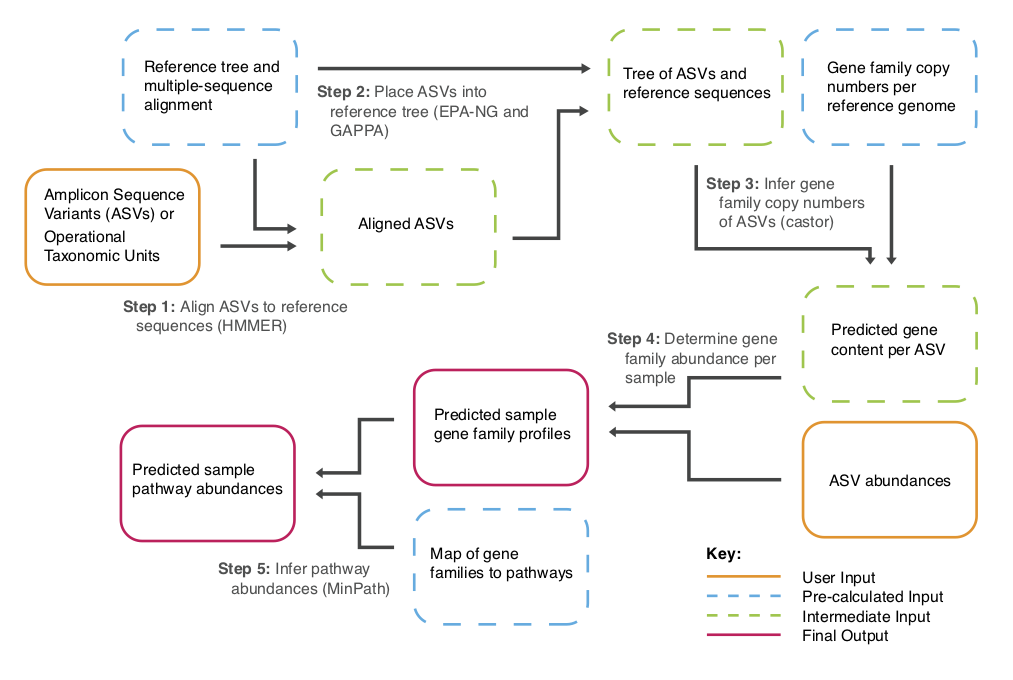
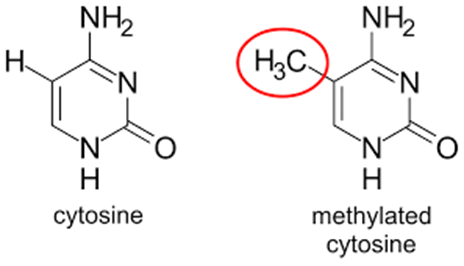
오늘은 균주 시퀀스 파일로부터 Antimicrobial Resistance genes 를 찾아내는데 사용할 수 있는 AMRFinder (AMRFinderPlus) 에 대해 간단하게 소개하려고 합니다. 이 프로그램을 사용하면 nucleotide sequence file 또는 protein sequence 를 .fa 형식으로 넣어주면 항생제 저항성 유전자들을 확인할 수 있으며 구동이 쉽고 어쨋든 이런 프로그램을 사용할때 가장 중요한 점은 얼마나 최신화된 데이터베이스를 기반으로 서치해주는지인데 NCBI DB 와 BLAST 를 기반으로 찾아주기 때문에 높은 신뢰도의 결과를 얻을 수 있습니다. 또한 이 툴은 뉴클레오타이드 또는 단백질 시퀀스에 point mutation 등 변형된 시퀀스에 대한 데이터도 포함하고 ..