그룹 간의 feature 의 상대적 풍부도를 비교하기 위한 도구로 LEfSe 를 사용하고 있는데요, LEfSe 관련 포스팅에서 공부했듯이 간단하게만 작동 원리를 알고 있었습니다. 그런데, 최근에 LEfSe 를 사용하다가 통계적으로 유의하게 나오는 것들에 대해 의구심이 생겨서 조금 더 자세히 직접 사용하는 코드와 함께 살펴보고자 포스팅합니다.
차원축소 (PCA, PCoA, LDA) in microbiome data 와 LEfSe : 2 (microbiome)
지난번 글에 이어서 LDA와 LEfSe에 대해 작성하겠습니다. # statquest 유튜브를 참고하였습니다. 3. LDA (Linear discriminant analysis) 앞서 살펴봤던 PCA 와 PCoA와 유사한 방법인 것처럼 보이지만, LDA는 지도학
hiimgood.tistory.com
LEfSe 코드 살펴보기
저는 QIIME2 를 사용하여 주로 분석하기에, QIIME2, 우분투 환경 내에서 LEfSe 를 사용, 분석할 수 있는 코드를 사용하고 있습니다. 아래 QIIME2 커뮤니티의 글을 참고하여 사용하고 있습니다.
LEfSe after QIIME2 to test at all taxonomic levels
Hello Akiriti, I was running into this issue as well after following the same steps listed [there] (Lefse after QIIME2) and collapsing the table down to level 6 If I am not mistaken (would love to have a moderator/admin/power user verify or correct my unde
forum.qiime2.org

(파라미터 이름들이 직관적이지 않아서, 먼저 위의 설명들을 보고 시작하시는게 좋습니다.)
1. prepare-lefse
$ dokdo prepare-lefse \
-t data/moving-pictures-tutorial/table.qza \
-x data/moving-pictures-tutorial/taxonomy.qza \
-m data/moving-pictures-tutorial/sample-metadata.tsv \
-o output/Useful-Information/input_table.tsv \
-c body-site \
-u subject \
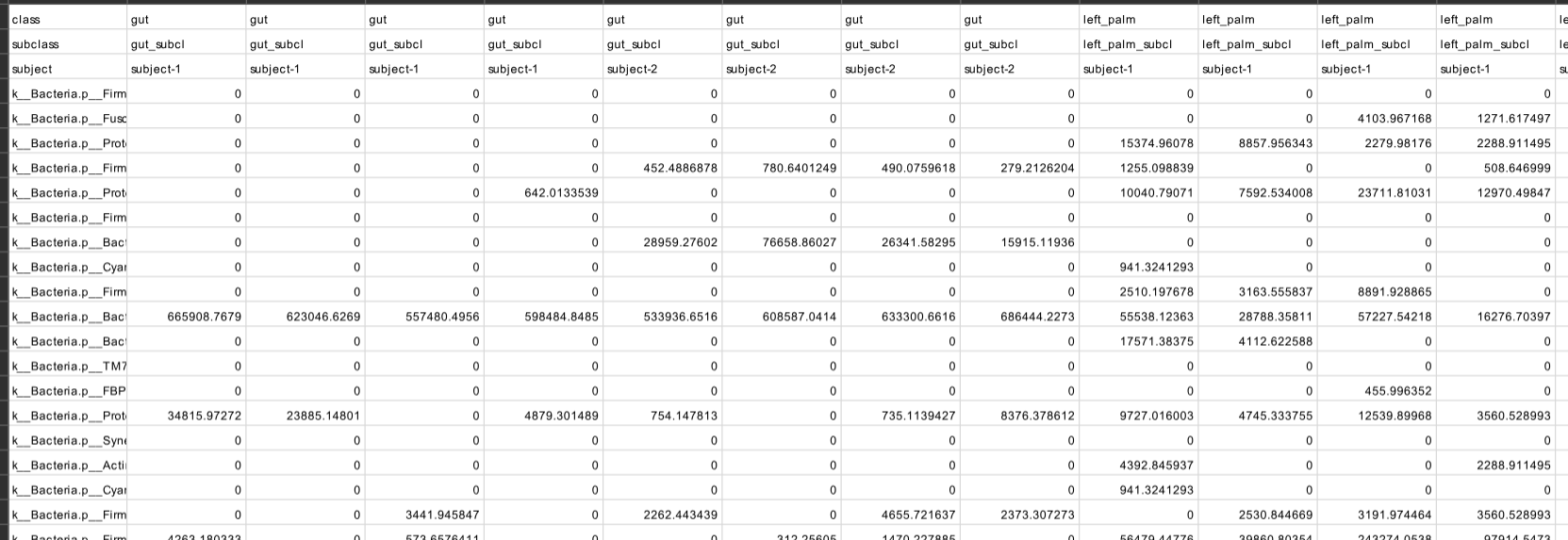
-w "[body-site] IN ('tongue', 'gut', 'left palm')"가장 먼저, lefse 를 수행하기 위한 input table 을 제작하는 단계입니다. (QIIME2 의 "Moving Pictures" 튜토리얼을 바탕으로 작성되었습니다.)
1. 일단 -t, -x, -m 는 위에서 넣어준 파일의 이름을 보면 알 수 있으므로 넘어가겠습니다.
2. -o 는 결과 파일의 위치와 이름을 설정합니다.
3. -c, -w 는 연결되어 있는 느낌이 있는데, -w 를 통해서 body-site 컬럼에서 필터링을 통해 분석할 class 를 선택합니다. 위의 코드에서는 tongue, gut, left plam 값을 갖는 것들만 사용하겠다는 의미이고, -c 는 -w 에서 사용한 컬럼명을 그대로 넣어줍니다. (여기서 -c 와 -w 의 컬럼명이 다르면 에러가 나므로 무조건 동일한 값을 줘야합니다.)
4. -u 는 subclass 입니다. 이건 코드 실행 결과 나오는 결과 파일을 보면 조금 더 빠르게 이해할 수 있는데, 1열의 body-site 로 필터링한 후에, 2열의 subject 를 기준으로 나눠준 모습입니다.

여기서 class, subclass 의 개념은 위의 분석 과정을 보면 어느 정도 이해가 갑니다. 모든 값을 대상으로 Kruskal-Wallis를 수행하고, 유의한 feature 에 한해서 subclass 로 묶어서 Wilcoxon 을 수행합니다.

(subject-1 값을 갖는 열이 왜 많은지 조금 의아할 수 있는데 위의 메타데이터를 확인하시면 연도별 샘플링을 진행해서 그렇습니다.)

이렇게 하면 lefse 를 돌리기 위한 첫번째 준비 파일이 완성됩니다.
2. lefse-format_input.py
$ lefse-format_input.py \
output/Useful-Information/input_table.tsv \
output/Useful-Information/formatted_table.in \
-c 1 \
-u 2 \
-o 1000000 \
--output_table output/Useful-Information/formatted_table.tsv여기서 다시 한번 lefse 를 돌리기 위한 두번째 준비가 필요합니다. 코드를 먼저 살펴보겠습니다.
여기서 먼저 알아야 할 것은 첫 줄의 input_table.tsv 만 넣어주면, 그 아래의 formatted_table.in 을 자동생성해 주고, formatted_table.tsv 가 결과파일로 나온다는 것입니다.
다 알아야 할 것은 -c, -u, -o 입니다.
-o 는 정규화를 위한 파라미터입니다. 아래의 formatted_table.tsv 를 보시면, 상대적 풍부도의 값이 상당히 커져있는 것을 볼 수 있습니다.
lefse 에서 정규화는 상대적풍부도의 값을 해당 샘플의 상대적 풍부도 합(=1) 로 나누고, -o 의 값을 곱해줌으로써 나온 값 입니다. 합이 1 이므로 쉽게 말하면, -o 의 값 (1000000) 을 곱해줬다고 생각하면 됩니다.
여기서 default 는 1,000,000 인데, 그 이유는 CPM normalization 을 기본으로 하기 때문입니다. CMP normalization 은
RNA-SEQ 에서 유전자의 발현량을 좀 더 보기좋은 숫자로 나타내기 위해 사용하는 것인데
CPM = count * 1,000,000 / total 입니다. (RNA-SEQ 에서 total reads 의 수가 매우 많기 때문에 1,000,000 을 곱해주지 않으면 너무 작은 수가 나오기 때문에 어느 정도의 보정을 위해 백만을 곱해줍니다.)
(솔직히 말하면 여기서 이 방법을 통해 정규화를 수행하는 이유를 잘 모르겠습니다... 기본적으로 값이 작아서일까요? 해당 값은 이미 relative abundance 로 정규화되었기에 total = 1 입니다. 그리고 값이 작은 것이 무슨 문제가 되는지도 잘 모르겠습니다... 우리는 발현량을 보고 싶은 게 아니라 통계적으로 유의한 지가 궁금한 것이므로 그 사이에 추가적인 연산을 통해 중간값들을 보기 좋게 만들어줘야 하는 이유를 잘 모르겠습니다. 찾아봐도 확실한 답을 찾지 못해서 관련해서 아시는 분들이 계시면 댓글이나 메일 주시면 너무 감사하겠습니다.)
+ -o 에 기본값인 1,000,000 일때 유의한 feature 가 잡히다가, 1 이나 1000 를 넣어주면 feature 가 잡히지 않습니다. 동일한 값을 곱해주는데 결과가 변하는게 신기합니다. 맞는건지 잘 모르겠어요
-c, -u 는 각각 class, subject 이 몇 번째 행인지 알려주기 위한 파라미터로, input_table.tsv 를 보시면 1행이 body-site, 2행이 subject 이므로 1, 2 값을 할당합니다.
3. run_lefse.py
$ run_lefse.py \
output/Useful-Information/formatted_table.in \
output/Useful-Information/output.res
# 결과
#Number of significantly discriminative features: 199 ( 199 ) before internal wilcoxon
#Number of discriminative features with abs LDA score > 2.0 : 199마지막입니다. 위에서 생성된 formatted_table.in 을 가지고 lefse 를 수행하여 output.res 파일을 얻게 됩니다.
(해당 res 파일을 가지고 lefse-plot_res.py, lefse-plot_cladogram.py 등을 수행하여 시각화도 가능하지만, 엑셀에 해당 파일을 드래그하면 raw 한 결과파일을 확인할 수 있습니다.)
지금까지 lefse 튜토리얼 코드를 가지고 하나씩 확인해 봤습니다. 저는 개인적으로 이번에 공부를 하면서 다른 프로그램 (aldex2, ancom-bc) 으로도 한번 분석해 봐서 결과를 비교해 봐야겠다고 생각했습니다. 뭔가 이해가 안 되는 부분이 있는데 그게 프로그램 자체의 문제인 건지 제가 아는 것이 많지 않아 이해를 못 하는 건지 알 수가 없어서 일단 다른 프로그램으로도 돌려보고 비교해 보는 게 최선인 것 같아요.
다음 포스팅에서 뵙겠습니다.
'bioinfo' 카테고리의 다른 글
| PERMANOVA in Beta-diversity (microbiome) (1) | 2023.11.27 |
|---|---|
| ANCOM-BC (Analysis of Composition of Microbiomes with Bias Correction) (2) | 2023.11.05 |
| QIIME2 feature classifier : 2 (naive bayes classifier) (1) | 2023.10.29 |
| enterotype (microbiome, R) (1) | 2023.10.28 |
| QIIME2 feature classifier : 1 (greengenes2 database) (1) | 2023.10.26 |