이번에 전사체 데이터 분석을 진행해 보면서 개인적으로 공부했던 부분들, 분석 과정 등을 하나씩 올려보려고 합니다. 원래 자주 하던 분석이 아니라 중간중간 틀린 부분이 있을 수 있는데 댓글로 수정해 주시면 바로 반영하겠습니다.
1. RNA-seq 과 transcriptome (전사체)
가장 가본적인 부분부터 천천히 공부해보려 합니다. 그러기 위해서는 central dogma 를 다시 한번 상기시켜볼 필요가 있습니다.
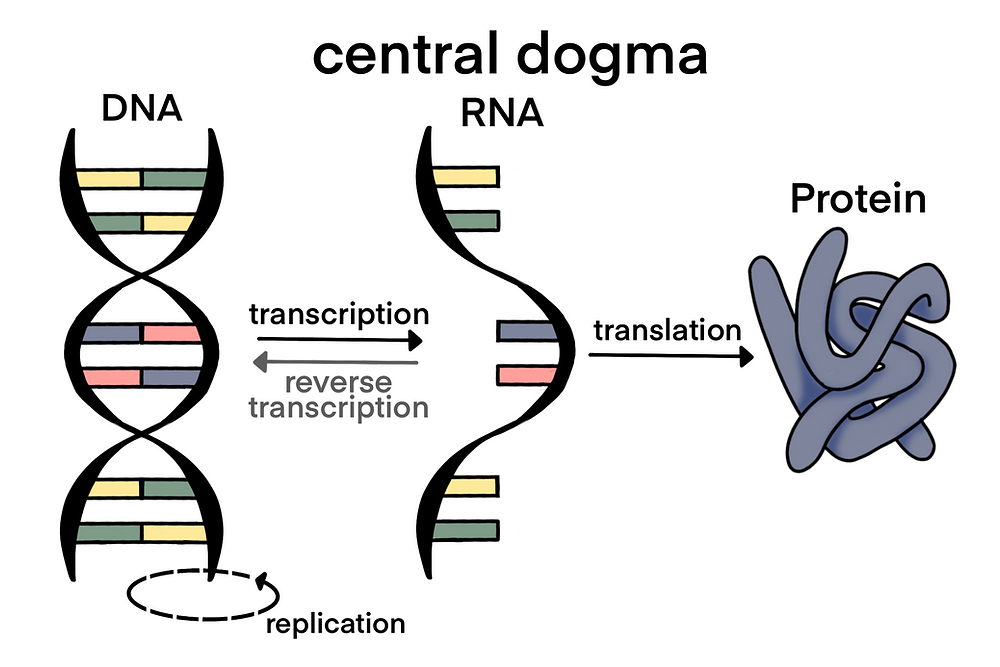
DNA 에서 RNA 를 합성하는 과정을 transcription (전사) 라고 합니다. 그러므로 transcriptome (전사체) 는 'mRNA 등을 포함하는 전사 과정에서 합성되는 물질들' 이라고 정의할 수 있습니다. 그럼 전사체를 시퀀싱, 분석한다는 의미는, 이 과정에서 합성된 물질 (mRNA 등) 의 유전정보를 확인한다는 의미입니다.
이것이 어떤 의미를 가질까요? DNA 와 RNA 에 존재하는 유전정보에 어떤 차이가 있는지를 알아야 합니다.
RNA 가 포함하고 있는 유전정보들은 DNA 의 유전정보를 기반으로 활성화된 유전자를 포함하는 첫 번째 매개체입니다. 그 말은, DNA 에 존재하는 모든 유전자가 항상 발현되는 것이 아니라 개체가 처한 상황에 따라 선택적으로 발현되거나, 그 발현량이 상향, 하향 조절되는데 그것을 RNA 를 통해 단백질을 합성하는 단계로 진행되고 우리는 그 과정의 가장 앞에 있는 RNA 를 분석하여 개체가 특정 환경에서 어떻게 반응하는지, 어떤 약물을 투여했을때 어떤 반응이 나타나는지 등에 대해 발현량을 수치적으로 확인할 수 있습니다.
2. 전사체 분석 기법
최근에는 특이한 상황이 아니라면, 대부분 RNA 를 추출하여 cDNA 를 합성, NGS 로 시퀀싱을 진행하는 것으로 알고 있습니다. 다만, 이 관련해서 찾아보면 항상 DNA microarray 에 대한 이야기가 항상 있더라구요. 그래서 간단하게 정리하고 넘어가겠습니다.
2-1. DNA microarray (DNA chip) technique
유리 슬라이드와 같은 고체 표면에 특정 유전자 서열을 탐지할 수 있는 형광표지된 DNA probe 를 부착합니다. 그리고 cDNA 가 해당 probe 에 결합하는 과정에서 나타나는 형광을 측정함으로써 RNA 상의 유전자 발현량을 정량하게 됩니다.
이 과정에서 두 그룹 간의 유전자 발현 차이를 확인하고자 할때는 두 그룹의 cDNA 를 서로 다른 형광으로 표지하면, 특정 DNA probe 에 결합할 때 나타나는 형광의 강도를 서로 비교할 수도 있습니다.
다만, DNA probe 를 제작하는 과정이 필요하기 때문에 우리가 정보를 갖고 있는 유전자 또는 개체에 대해서만 적용이 가능하다는 한계가 있으며 실험에 필요한 RNA 의 양이 상대적으로 (NGS 에 비해) 많은데 RNA 는 불안정하고 많은 양 추출이 어렵기 때문에 지금은 NGS 를 통해 RNA (cDNA) 의 서열을 확인하는 방법을 일반적으로 사용합니다.
2-2. NGS (Sequencing using cDNA)
NGS 부분은 Illumina 기준 다른 시퀀싱 기술들과 동일한 기법을 사용합니다. (flow cell 에 결합 이후 bridge amplification) 다만, RNA 를 그대로 시퀀싱에 사용하는 것이 아니라, RNA 추출 이후 상보적인 complementary DNA 를 합성한 후 시퀀싱 진행합니다.
3. RNA-seq 데이터 분석 과정

일반적인 RNA-seq 데이터 분석 과정입니다. 추출한 RNA 에 대한 시퀀싱 데이터를 전달받으면, 시퀀싱 데이터에 대한 퀄리티를 확인하고 (FASTQA 등) 시퀀싱 과정에서 사용한 adapter 를 제거 (Trimmomatic 등) 해줍니다.
QC 가 완료된 reads 를 reference genome 에 mapping 하는 과정을 수행합니다. (STAR 등)
reference data 에 alignmnet 하는 도구들은 상당히 많은데 어떤 도구를 사용했는지에 따라 성능에 차이가 있다고 합니다.
완성된 유전자 데이터를 전사체 데이터로 변환하고 유전자 발현의 차이를 정량하는 과정을 거쳐 어떤 유전자가 어느 샘플에서 더 많이, 혹은 더 적게 발현되었는지 유의한 유전자를 확인하는 DEG (differentially Expressed Gene) 분석을 진행합니다. (DESeq2, edgeR 등)
다음 포스팅에서는 reference genome 데이터를 선택하는 과정부터 알아보겠습니다,
'[RNA-seq]' 카테고리의 다른 글
| RNA-seq 분석 : 3. read 의 mapping 및 normalization (0) | 2023.11.20 |
|---|---|
| # log2 fold change (1) | 2023.11.17 |
| RNA-seq 분석 : 2. reference genome 의 종류 (1) | 2023.11.16 |