이번에 전사체 데이터 분석을 진행해 보면서 개인적으로 공부했던 부분들, 분석 과정 등을 하나씩 올려보려고 합니다. 원래 자주 하던 분석이 아니라 중간중간 틀린 부분이 있을 수 있는데 댓글로 수정해 주시면 바로 반영하겠습니다.
이전 포스팅에서와 같이 필요에 따라 알맞은 형태의 reference genome 을 얻으면 그 위에 시퀀싱을 수행하여 얻은 수 많은 reads 들을 maping 하는 과정이 필요합니다. 이 과정은 일반적인 alignment 과정과 다르지 않고 일반적으로 프로그램을 통해 수행되기 때문에 그 과정에 대해서는 소개하지 않겠습니다. (HISAT2, STAR, Bowtie2 등)
수행하고 나면 reference genome 상에 우리의 reads 들이 mapping 되는데, 우리의 목표는 어떤 유전자가, 얼마나 많이 혹은 적게 발현되는지를 확인하는 것 입니다.
그래서 먼저, reads 가 위치하는 곳이 어떤 유전자에 해당하는지를 먼저 알아야 합니다. 이전 포스팅을 보시면 genome fasta file 말고도 gene annotation (GFF) file 까지 다운로드하였습니다.
GFF (General Feature Format) 형식은 아래의 ensemble 페이지에 잘 설명되어 있습니다. 아래의 사진과 같이 염기서열의 위치, 방향, 해당 염기서열이 어떤 유전자에 해당하는 지 등에 대한 annotation 정보가 담겨있는 파일입니다.
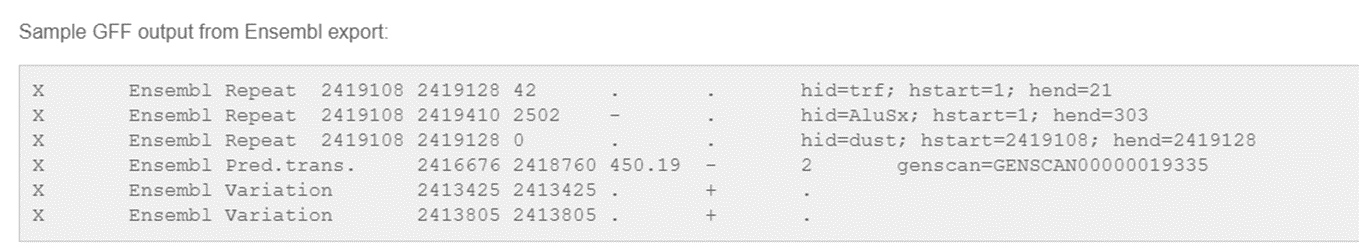
해당 GFF filte 과 mapping 된 파일을 사용하여 어떤 gene 또는 transcript 에 몇 개의 reads 가 붙었는지 확인할 수 있습니다. 그럼 여기서 확인된 reads 의 개수를 이후 분석에 그대로 사용한다면 어떻게 될까요?

그렇게 되면 몇 가지의 편향이 존재할 수 있습니다.
1. 일반적으로 생각해보면 (동일한 발현량을 갖는다고 가정할 때) 길이가 긴 유전자는 길이가 짧은 유전자보다 더 많은 reads 에 의해 mapping 될 확률이 높습니다.

동일하게 1의 발현량을 갖는다고 가정하면 길이가 길수록 더 많은 reads 가 매핑될 것 입니다. (물론 이렇게 정직하게 매핑되진 않겠지만 단순하게 생각해보면 그렇습니다.)
2. 샘플 마다 생성되는 total sequencing reads 의 개수 또한 다릅니다. 이런 것들을 고려하면, reads 수를 그대로 비교하는 것은 편향을 포함하게 되어 정상적인 비교가 이루어지지 않습니다.
3. 또한 시퀀싱 샘플에 따라 sequencing depth 의 차이 등도 다르기 때문에 이러한 시퀀싱 과정에서의 기술적인 차이들도 고려해야 합니다.
하여, reads 개수를 바로 사용하지 않고 정규화하여 사용하게 되는데 여러 가지 정규화 기법들이 있습니다.
1. CPM (counts per million) mapped reads
이는 가장 간단한 정규화 기법으로, 위에서 언급한 편향 중에서 2. 샘플 간의 total sequencing reads 개수의 차이를 보정할 수 있습니다. 그러나 이는 위의 1번 편향을 보정해주지 못합니다.
2. RPKM, FPKM (Reads / Fragments per kilobase of trasncript per million Reads / Fragments mapped)
RPKM = CPM * 1,000 / gene length 입니다.
FPKM 과 RPKM 은 유사한 개념으로 시퀀싱 과정에서paired-end 를 사용했다면 FPKM / single-end 를 사용했다면 RPKM 을 사용합니다.
위 기법은 CPM 에 추가로 유전자 길이의 차이에 대하여 보정해줍니다. 따라서 1, 2번 편향에 대해 보정해줄 수 있는 정규화 방법으로 주로 사용되고 있습니다.
간단하게는, 아래와 사진과 같이 정리할 수 있습니다. (보정을 위한 1,000,000 / 1,000 을 곱하는 과정을 제외)
다만 이 방법을 사용했을 때 나타나는 한 가지 문제점은 샘플 간에 정확한 비교가 어렵다는 것 입니다. 어떻게 보면 마이크로바이옴 데이터를 relative abundance 로 나타냈을 때와 비슷한 문제인데요,
A 샘플의 E. coli 가 0.4 / B 샘플의 E. coli 가 0.4 의 relative abundane 로 존재할 때, 두 샘플 안에 E. coli 가 동일한 양으로 존재한다고 말하지 못하는 것과 유사합니다.
3. TPM (transcripts per million)
TPM = RPKM / Total RPKM * 1,000,000 입니다.
이는 샘플 간 유전자 양의 비교를 용이하게 하기 위해 만들어졌습니다. 계산식을 보면 알 수 있듯이, RPKM 에 대한 relative abundance 를 계산한 것 같이 보이기도 합니다.
그러면 위에서 E. coli 의 비교를 봤을때, 샘플 내의 합이 1이 되는 것이 아니라 샘플 간의 E. coli 양의 합이 1이 되는 것과 동일한 효과를 낼 수 있습니다. -> 1, 2, 3 번의 편향에 대해 보정해준다고 할 수 있습니다.
다만, 이 방법도 많이 사용되지는 않습니다. 어쨌든 RPKM 을 기반으로 계산하다 보니 샘플 간의 비교를 정확하게 하기 위해서는 TMM normalization, Quantile 등 다른 방법을 사용하는 것이 바람직하다고 합니다.
# reference
'[RNA-seq]' 카테고리의 다른 글
| # log2 fold change (1) | 2023.11.17 |
|---|---|
| RNA-seq 분석 : 2. reference genome 의 종류 (1) | 2023.11.16 |
| RNA-seq 분석 : 1. 기본개념 (0) | 2023.11.14 |