다양성 분석을 수행하기 전에 representative sequence 로 부터 phylogeny 를 만들어야 합니다. 이를 통해 rep-seqs 의 계통수를 계산하고 rooted tree 를 기반으로 Unifrac metrics 등을 생성할 수 있습니다. 해당 부분에 대해 qiime2 코드와 함께 알아보겠습니다.
1. Representative sequences alignment
phylogeny 를 만들기 전에 multiple sequence alignment (MSA) 를 생성해야 합니다. MSA 란, 다수의 서열의 공통된 부분을 찾아 정렬하는 것입니다.
MSA 는 생물학적 서열의 진화 분석을 하는데에 중요한 역할을 하며 QIIME2 에서는 MAFFT (Multiple Alignment using Fast Fourier) 라는 알고리즘을 사용하여 MSA 를 생성하며 MAFFT 에 대한 내용은 아래 논문에서 조금 더 자세하게 확인할 수 있습니다.
MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability
Abstract. We report a major update of the MAFFT multiple sequence alignment program. This version has several new features, including options for adding unalign
academic.oup.com
우리의 경우에는 주로 V4 region 의 300bp 정도 되는 representative sequences 를 정렬하여 이 정보를 바탕으로 phylogeny 를 만들게 됩니다.
Qiime2 를 통하여 수행하는 코드는 다음과 같으며, 결과 파일로 정렬된 rep-seqs 를 반환합니다.
qiime alignment mafft \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza
해당 결과 파일을 .qzv 로 변환하여 확인해보면 다음과 같습니다.
(mafft 수행 전 rep-seqs)

(mafft 수행 후 aligned-rep-seqs)

MSA matrix 에서 각 열은 상동성 관계에 있는 뉴클레오타이드 임을 의미합니다. 만약, 한 시퀀스의 A 와 다른 시퀀스의 G 가 동일한 열에 있다면 그 두개의 뉴클레오타이드가 반드시 하나의 공통된 조상 뉴클레오타이드에서 일련의 치환 또는 대체를 통해 진화하다는 것을 의미합니다. '-' 는 indel (insertions and deletions) 입니다.
2. Masking representative sequences alignment
마스킹은 계통 분석을 수행하기 전에 유용하지 않거나 애매한 정렬 오류를 제거하기 위해 수행합니다. 많은 정렬 오류가 노이즈를 일으키고 계통적 추론을 하는데에 있어 방해가 되기 때문에 이러한 과정은 필수적입니다.
이에 MAFFT 로 정렬된 파일에서 일부 모호하게 정렬된 부분을 제거합니다. QIIME2 코드는 아래와 같습니다.
qiime alignment mask \
--i-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza
(마스킹 수행 후 masked-aligned-rep-seqs)


3. Construct a phylogeny
이제 완료된 MSA 를 기반으로 계통발생을 예측합니다. 많은 도구들이 있지만 여기서는 FastTree 파이프라인을 사용합니다. (계통발생을 예측하는 많은 도구들이 있는데 해당 부분에 대해서는 나중에 포스팅하도록 하겠습니다.)
qiime phylogeny fasttree \
--i-alignment masked-aligned-rep-seqs.qza \
--o-tree fasttree-tree.qza
(해당 fasttree-tree.qza 를 시각화하기 위해서는 QIIME2 에서는 불가하고 ITOL (interactive tree of life) 라는 사이트에서 tree.qza 파일을 업로드하고 MSA qza 파일을 드래그, 드롭 함으로써 수행할 수 있습니다.)

4. Root the phylogeny
Unifrac 과 같은 diversity metrics 를 적절하게 만들기 위해서는 phylogeny 가 rooted 되어야 합니다. phylogenetic tree 를 rooting 한다는 것은 하나의 특정 노드를 뿌리(root) 라고 불리는 특별한 노드로 지정되고 트리에 있는 다른 가지들은 뿌리로부터 멀어지는 방향으로 이어져나갑니다.
rooting 이 중요한 이유는 가장 먼저 그림을 조금 더 직관적으로 나타내기 위함에 있습니다. 이것은 생각보다 중요합니다. 위의 unrooted-tree.qza 를 시각화한 그림과 아래 우측의 Mid-point rooted 된 그림을 비교해 보면 기본적으로 복잡도에 차이는 있지만 아래의 그림이 직관적으로 이해하기 쉬움을 알 수 있습니다. 또한, 진화의 방향과 관련성에 관해 추론하기 위해서는 올바른 뿌리가 있어야 합니다. Unrooted tree 에서는 진화의 방향을 확인할 수 없기 때문에 이를 해석하고 사용하기 어렵습니다.
Rooting 을 위한 방법에는 Midpoint rooting, Outgroup rooting 이 있는데 QIIME2 에서는 Midpoint rooting 방법을 사용합니다.
Midpoint rooting 은 아래 사진과 같이 중간 지점을 root node 로 지정하는 것입니다.
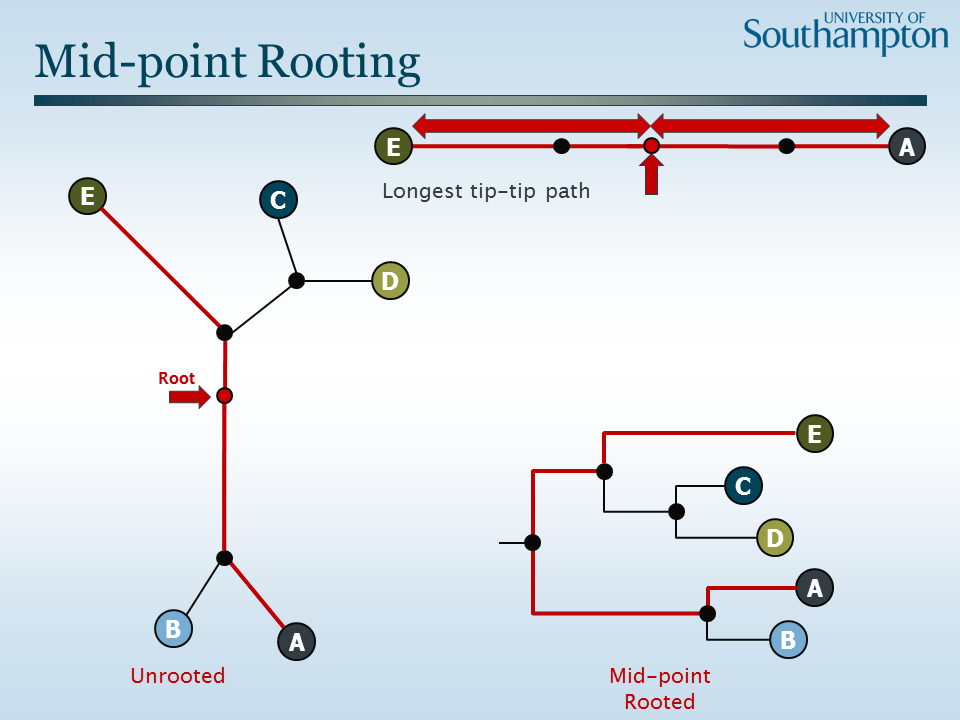
지금까지 rep-seqs 에서 unrooted tree 까지 diversity 분석을 위한 전처리 과정에 대해 알아보았습니다. 여기서는 한 단계마다 하나씩 코드를 작성하였지만 실제로는 MSA 부터 unrooted tree 까지 하나의 코드를 사용하여 한번에 결과를 얻을 수 있습니다. 아래에 코드를 첨부하니 실제 분석에서는 아래의 코드를 사용하는 것이 훨씬 시간을 단축할 수 있습니다.
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--output-dir mafft-fasttree-output
# reference
1. https://academic.oup.com/mbe/article/30/4/772/1073398?login=false
2. https://docs.qiime2.org/2022.8/tutorials/phylogeny/?highlight=phylogeny#fasttree
3. https://cabbagesofdoom.blogspot.com/2012/06/how-to-root-phylogenetic-tree.html
'bioinfo' 카테고리의 다른 글
| 인코렌탈 체험, 마이크로바이옴 데이터 간단 분석 (CLC Genomics Workbench Premium , incoRENTAL) (1) | 2024.04.22 |
|---|---|
| DNA Methylation 탐지를 위한 시퀀싱 기법 (0) | 2024.04.07 |
| Annotation (RAST) (0) | 2023.12.28 |
| Unifrac distance (microbiome) (0) | 2023.12.16 |
| PERMANOVA in Beta-diversity (microbiome) (1) | 2023.11.27 |