저는 NGS data 를 시퀀싱 업체에서 받으면 데이터의 전처리를 위해 DADA2 를 사용합니다.
Divisive Amplicon Denoisinig Algorithm (DADA2)
DADA2: High resolution sample inference from Illumina amplicon data
We present DADA2, a software package that models and corrects Illumina-sequenced amplicon errors. DADA2 infers sample sequences exactly, without coarse-graining into OTUs, and resolves differences of as little as one nucleotide. In several mock communities
www.ncbi.nlm.nih.gov
DADA2 는 대표적인 시퀀싱 데이터에 대한 quality control 을 위한 프로그램입니다. (quality filtering, chimera checking, paired-end read joining 등)
논문에 의하면,
DADA2 는 Illumina 기기를 사용하여 시퀀싱 된 데이터를 전처리하는 데에 자주 사용되는 프로그램 패키지입니다. 이전의 OTUs 를 기반으로 클러스터링 하던 프로그램들과 달리, ASVs 를 기반으로 샘플의 서열을 클러스터링, 식별하기 때문에 더 좋은 정확도와 식별능을 갖는 것이 특징입니다. 추가로, 해당 프로그램은 PCR, sequenicng 과정에서 발생하는 노이즈를 식별, 제거하는 데에도 뛰어나 더욱 높은 품질의 분석 결과를 얻을 수 있습니다.
라고 합니다.
이번 포스팅에서는 DADA2 프로그램의 이론적인 배경보다는 실제 프로그램을 사용하기 위해 필요한 input file, 분석 결과 받을 수 있는 output file 에 대해 자세히 알아보고자 합니다.
DADA2 는 제가 알기로 R 과 qiime2 등과 연결해서 주로 사용하는 것으로 알고 있는데 저는 qiime2 에서 주로 구동하므로 해당 방식에 대해서 알아보겠습니다.
qiime dada2 denoise-paried
--i-demultiplexed-seqs 'demultiplexed_input.qza' /
--p-trim-left-f 5 /
--p-trim-left-r 4 /
--p-trunc-len-f 230 /
--p-trunc-len-r 235 /
--output-dir DADA2가장 일반적으로 qiime2 에서 dada2 를 실행하는 코드입니다.
input 파일로는 pooled 된 샘플들을 demux 실행한 후의 데이터를 넣어줍니다.
각 파라미터들은 명령어가 상당히 직관적이어서 금방 알아볼 수 있는데요,
trim-left : 5' 부터 몇 번째 뉴클레오타이드까지 자를 것인지
trunc-len : 반대쪽 (3'방향) 의 몇 번째 뉴클레오타이드까지 사용할 것인지
-f : forward (r1)
-r : reverse (r2)
output-dir : output 파일 저장할 장소
입니다.
시퀀싱한 데이터를 어디까지 자르고, 어디까지 사용할 것인지는 직접 quality score 를 보면서 설정하게 되는데요, 다소 주관적인 부분이기도 합니다. 이것과 관련된 포스팅들은 많이 해놓았습니다. (phred score, sequencing quality (궁금했던 것들) 등)
그중 한 개 포스팅만 링크 달아놓겠습니다. 참고하시면 좋을 것 같아요.
Sequencing quality (궁금했던 것들) : 1 (illumina Miseq)
오늘은 실제 NGS 데이터의 QC를 수행하면서, 궁금했던 부분에 대해 공부한 것을 작성하기 위해 포스팅합니다. Sequencing quality (Phred quality score) : 1 (microbiome) 시퀀싱이 완료된 후 파일을 전달받아서 d
hiimgood.tistory.com
이제 위와 같은 코드를 실행시키게 되면 DADA2 파일에 몇 개의 output 파일들이 생성될 것입니다.
위에서 말씀드렸다시피, 저는 qiime2 를 통해 구동했으므로 qiime2 에서 dada2 를 수행하면, 일반적으로 3가지의 파일을 얻을 수 있습니다.
1. feature table
2. representataive sequences (rep_seqs)
3. denoising stats
1. feature table
qiime2 특성상, qza 파일을 qzv 로 변환하면 qiime2 view 를 통해서 쉽게 시각화할 수 있습니다.
지금부터 보여드리는 파일의 시각화들은 모두 qzv 변환을 통해 시각화한 것들입니다.

feature table 파일을 요약한 것입니다. 위에서 dada2 는 ASV 를 기반으로 시퀀싱 데이터를 클러스터링 한다고 말씀드렸었습니다. 위의 요약은 demux 된 샘플의 수, ASV 의 수에 관한 정보입니다.
아래의 Frequency per sample 을 보면, 위 시퀀싱 데이터에서는 샘플 당 평균 약 29535 개의 ASV 를 갖고 있으며, 최소 ASV 를 갖는 샘플은 9935 개, 최대 약 56000 개의 ASV 를 갖는 샘플도 있습니다.
이 요약 데이터를 갖고 이후 sequencing depth 를 어떻게 설정할 것인지를 결정할 수 있습니다.
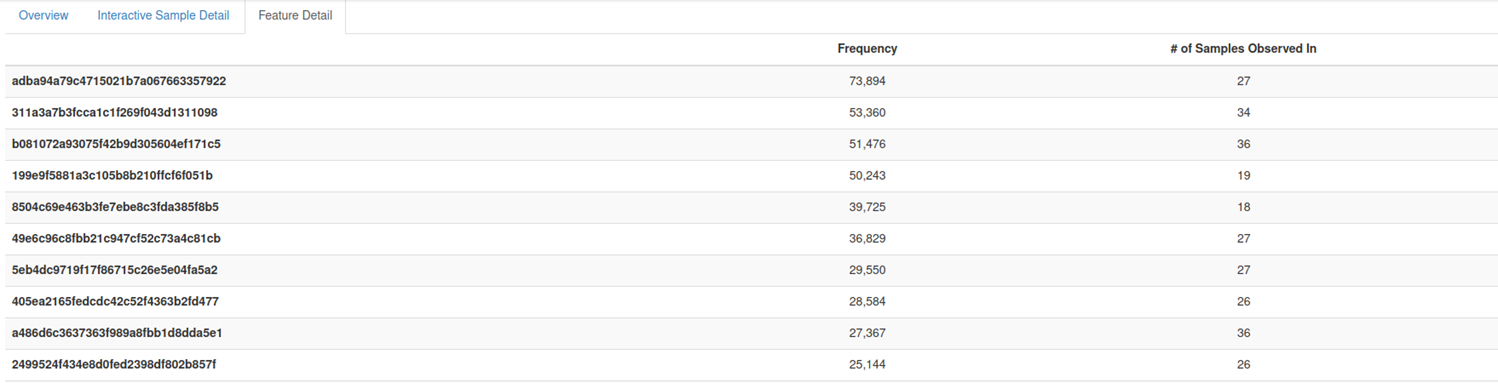
feature Detail 을 확인하면 이렇게 각 ASV 가 몇 번 나타났는지, 몇 개의 샘플에서 확인되었는지를 볼 수 있습니다.
저렇게 알파벳과 영어가 합쳐진 것이 각 ASV 의 ID 를 나타내는데 hash 라고 하는 기법을 사용해서 고유하게 식별할 수 있도록 합니다.
2. representataive sequences (rep_seqs)

우리는 ASV 에서 representative sequence 라는 개념에 대해 이미 배웠습니다. 위 feature_table 파일에서 해시기법을 통해 생성된 하나하나의 rep_seqs 에 대한 정보를 확인할 수 있는 파일이 '2. representataive sequences (rep_seqs)' 입니다.
statistics 를 보면, 모든 read 를 동일한 파라미터로 trimming 했기 때문에 240 의 길이를 동일하게 같는 것을 볼 수 있습니다.
그리고 아래의 sequence table 에 보면 각 rep seqs 를 NCBI database 에 검색해서 정보를 확인할 수도 있으며 fasta file 형태로 다운로드하여 사용할 수 있습니다.
3. denoising-stats
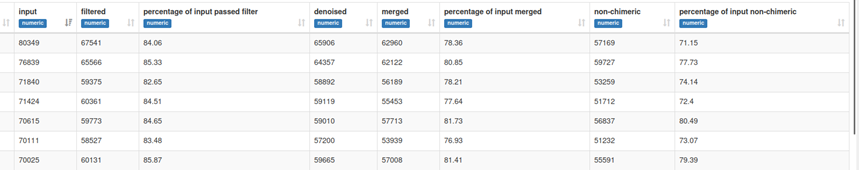
(가장 왼쪽에 잘린 부분은 sample-id 입니다)
제목 그대로 sequencing read 에 대한 quality control 이후의 수치적인 부분을 확인할 수 있는 파일입니다. 예를 들어, 가장 위의 샘플을 보면, 최초 80349 개의 reads 에서 시작해서, 모든 필터링을 거치고 나서 최종 57169 개의 reads 가 남았습니다.
(paired-end 라는 것은 r1 read 와 r2 read 를 겹쳐서 만들어지기 때문에 80349 개의 read 가 의미하는 것은 r1 read 개수 또는 r2 read 개수와 동일합니다.)
1. filtered : Q score 를 기반으로 한 filtering 을 수행합니다.
2. denoised : denoising algorithm 을 기반으로 filtering 수행합니다. 알고리즘은 아래와 같습니다.
denoising algorithm 에서는 오류 모델을 사용해서 개별 read 에 대해 독립적인 검증을 통해 error read 를 찾아냅니다.이 오류 모델에 대해서는 이해하기 쉽지 않아 제가 이해한 선에서 정리해 보겠습니다.
"""
오류 모델은 포아송 분포를 기본으로 합니다. 포아송 분포는 '단위 시간 안에 어떤 사건이 몇 번 발생할 것인지를 표현' 합니다. 따라서, 단일 뉴클레오타이드의 전환, 삭제, 삽입 등의 mutation 이 일어날 가능성에 대해 계산함으로써 이를 오류 모델로 나타냅니다.
위 아이디어를 바탕으로 예시를 들어보면,
AAAAA 라는 read 에서 AAAAG 로 마지막 염기가 바뀔 확률을 오류 모델을 바탕으로 계산했더니 20% 였다고 가정합니다.전체 read 를 살펴보니 AAAAA 가 2개, AAAAG 가 3개였습니다.
그런데 AAAAG 가 정상적인 read 가 아닌 시퀀싱으로 생성된 오류 read 라면,총 5개의 AAAAA + AAAAG 의 구성이 2개, 3개가 아닌, 4개, 1개 로 나타나야 합니다.
그러므로 AAAAG 는 시퀀싱 중에 나타난 오류가 아닌 실제 존재하는 read 임을 확인할 수 있습니다.
"""
제가 이해한 부분을 가장 간단한 방식으로 설명한 것이어서 실제와는 조금 다를 수 있습니다. 사실 완전히 다를 수도 있습니다. ㅜ 그런 경우에는 댓글 남겨주세요.
3. merged : merged 이후에 남은 시퀀스 개수 (이를 통과하지 못했다는 것은 read 가 너무 짧거나 겹치지 않아 merge 하지 못했다는 것을 의미)
4. non-chimeric : chimera 는 PCR 과정에서 인위적으로 생성된 DNA 서열입니다. 자연에 존재하지 않는 서열을 제거하는 과정입니다.
이렇게 총 4개의 과정을 거쳐 최종 feature table 을 구성할 read 의 수를 확인할 수 있습니다.
이상으로 DADA2 의 input, output 파일이 어떻게 구성되어 있는지에 대해 알아봤습니다.
# reference
1) Callahan, Benjamin J., et al. "DADA2: High-resolution sample inference from Illumina amplicon data." Nature methods 13.7 (2016): 581-583.
'bioinfo' 카테고리의 다른 글
| enterotype (microbiome, R) (1) | 2023.10.28 |
|---|---|
| QIIME2 feature classifier : 1 (greengenes2 database) (1) | 2023.10.26 |
| Sampling depth 설정 관련 (1) | 2023.10.08 |
| Sampling depth 와 rarefaction (microbiome) (2) | 2023.10.07 |
| Data augmentation in microbiome data (데이터 불균형) (0) | 2023.09.06 |