이전 글에서 말했듯이 DADA2 를 수행하고 나면, representative sequence 정보를 얻을 수 있습니다.
그 다음에 해야 할 일은, 해당 rep seqs 이 어떤 미생물에 해당하는지 확인해야 하는 작업입니다.
기본적으로 저는 EMP 의 16S Illumina Amplicon Protocol 을 따라 시퀀싱 하였습니다. 해당 프로토콜에서는 16S 유전자의 V4 region 을 증폭시켜 시퀀싱 수행하며, 515F-806R primer 를 사용합니다. 따라서 오늘 포스팅할 데이터베이스 관련해서는 total 16s rRNA gene 또는 whole genome shotgun sequencing 과는 사용할 데이터베이스가 다르므로 참고만 해주시면 될 것 같습니다.
일단 데이터베이스 관련해서 개인적인 배경을 조금 더 말씀드리면, 저는 qiime2 를 사용해서 데이터분석을 진행하는데 특히, taxonomic assignment 를 위해 Naive Bayes classifier 를 사용하고 있습니다. (세부적인 부분은 추후에 포스팅 할 예정입니다.) qiime2 에서 16S rRNA 데이터 분석을 위해 NB classifier 을 사용할 때 사용 가능한 데이터베이스는 두 개가 있는 것으로 알고 있습니다. 제목에 나와있는 것 처럼 SILVA database 와 greengenes 가 그것들입니다.
두 데이터베이스가 가지고 있는 데이터에 조금의 차이가 있는데, 16S rRNA 데이터는 두 데이터베이스 모두 가지고 있기 때문에 어떤 것을 사용할지는 그렇게 중요한 부분은 아닌 것으로 알고 있습니다. 다만, 저는 SILVA database 를 주로 사용했었는데 그 이유 중 제일 큰 것이 SILVA 는 주기적인 업데이트를 해준다는 것입니다. greengenes 는 제가 알기로 2013 년쯤 업데이트 이후 추가적인 업데이트가 없었던 것으로 알고 있습니다. 그래서 아무래도 계속해서 데이터가 추가되는 SILVA 를 사용했었습니다.
SILVA database : https://www.arb-silva.de/

홈페이지를 자주 들어가지는 않지만 깔끔하게 잘 꾸며놨습니다. (greengenes 는 따로 홈페이지는 없는 것 같습니다. 데이터베이스를 바로 다운로드하게 되어있고 꾸며놓지는 않았더라구요)
그런데 최근에 greengenes 2 가 나오면서 논문이 출판되었는데 읽어보니 greengenes 2 가 상당히 좋아진 것 같아 넘어가게 되었습니다. 이번 포스팅에서는 해당 논문을 간단하게 보면서 qiime2 코드까지 확인해보겠습니다.

Greengenes2
greengenes2 의 기본 아이디어는 기존의 단점을 보완하기 위해 자주 사용되는 taxonomy, phylogeny database 들을 통합한다는 것입니다. (single massive reference tree 라고 표현합니다)
통합하는 과정에 대해 이해하기 위해서는 'uDance' 라는 프로그램에 대해 알아야 합니다. 저도 이 논문 보면서 알게 된 프로그램이라 정말 간단하게만 확인하였습니다. 조금 더 자세하게 보시고자 하는 분들은 아래 깃허브를 확인하시면 됩니다.
GitHub - balabanmetin/uDance
Contribute to balabanmetin/uDance development by creating an account on GitHub.
github.com
Udance
해당 프로그램을 설명할 때 가장 중요한 단어가 backbone tree 인 것 같은데요, 큰 하나의 phylogenetic tree 데이터를 기본으로 가지고, 새로운 sequence 데이터가 들어왔을 때, 그것이 새로운 것이면 새로운 가지를 만들고, 기존에 존재하는 것이라면, 그곳에 통합하게 됩니다. 자세한 알고리즘은 확인해보지 않았지만, 시퀀싱 과정에서의 bias, 16s rRNA gene 상의 돌연변이 (conserved region 인데 돌연변이가 자주 나타나는지는 잘 모르겠습니다) 같은 염기의 차이를 인지하여 그것이 단순한 오류 또는 돌연변이인지, 완전히 다른 종인지 확인한다고 합니다.
이런 과정을 거대한 데이터에 대해서 처리할 수 있는 프로그램이라고 하네요.
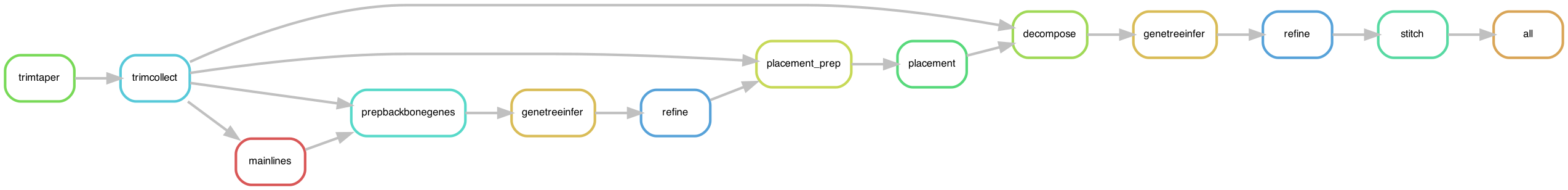
다시 greengenes2 로 돌아와서, greengene2 는 위의 uDance 를 사용해서 여러 데이터베이스를 통합한 형태입니다.
WoL1 (10,575 genes) / Living Tree Project (January 2022 released, 18,356 full-length 16s rRNA sequence) / Karst et a. (1,725,274 near-complete 16S rRNA genes) / data of EMP500 / GTDB r207 (all full-length 16S rRNA sequences) / inserted 23,113,447 short V4 16S rRNA Deblur v1.1.0 / amplicon sequence variants (ASVs) from Qiita (retrieved 14 December 2021) / mitochondria and chloroplast 16S rRNA from SILVA v138 using deep-learning-enabled phylogenetic placement (DEPP) v0.3 / ASVs from over 300,000 public and private samples in Qiita, including the entirety of the EMP13 and American Gut Project/Microsetta
정말 많은 데이터베이스를 통합했음을 알 수 있고, 그중에는 SILVA 도 포함되어 있어서 greengenes2 를 사용하는 게 더 유리해진 것 같아요.

각 데이터들이 어느 데이터베이스에서 가져온 것들인지 표현하는 그림입니다. 다만, legend 색을 연한 색을 일부러 썼는지는 모르겠지만 잘 보이지는 않네요.
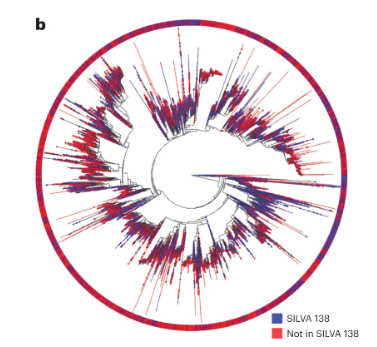
간단하게 논문의 그림 하나만 더 보면, greengenes2 데이터를 BLAST 했을 때 나온 best hit 을 가지고 SILVA 138 에 있는 데이터와 없는 데이터를 표현한 것이고 빨간색이 주를 이루는 것을 보아 확실히 greengenes2 가 더 많은 데이터를 갖고 있다는 것을 확인하였습니다. 이는 아마 더 세부적인 genus 들을 구분할 수 있다는 것이겠죠. (SILVA 뒤의 숫자는 버전입니다. 138은 2019년 버전이네요)
해당 데이터베이스를 다운로드하기 위해서는 아래의 사이트를 이용하시면 됩니다. 저의 경우에는 앞서 말씀드렸다시피, V4 region 증폭한 데이터를 사용해서, nb classifier 를 돌려 assignment 를 수행하기 때문에 2022.10.backbone.v4.nb.qza 를 사용합니다.
Index of /greengenes_release/2022.10-rc1
ftp.microbio.me
데이터베이스를 다운로드하면, 이제 qiime2 상에서 데이터베이스를 hashed 된 rep seqs 가 어떤 미생물인지 알 수 있게 됩니다. 해당 과정에 대해서는 다음 포스팅에서 이어가겠습니다.
# reference
1) McDonald, Daniel, et al. "Greengenes2 unifies microbial data in a single reference tree." Nature biotechnology (2023): 1-4.
2) https://forum.qiime2.org/t/introducing-greengenes2-2022-10/25291
'bioinfo' 카테고리의 다른 글
| QIIME2 feature classifier : 2 (naive bayes classifier) (1) | 2023.10.29 |
|---|---|
| enterotype (microbiome, R) (1) | 2023.10.28 |
| Divisive Amplicon Denoisinig Algorithm (DADA2) output file (microbiome) (1) | 2023.10.21 |
| Sampling depth 설정 관련 (1) | 2023.10.08 |
| Sampling depth 와 rarefaction (microbiome) (2) | 2023.10.07 |