마이크로바이옴 데이터를 분석할 때 자주 사용되는 분석 워크플로우는 있을 수 있지만 마이크로바이옴 데이터의 특성상 아직까지 정형화된 것은 없습니다.
데이터를 분석하는 데에 있어 다양한 도구들이 교차 사용되고 있으며 현재까지는 자신의 데이터 특성에 맞는 도구를 선택하여 시각화하고, 독자를 설득하는 방식으로 많은 분석이 진행되고 있습니다.
이와 같은 방식으로, 미생물군집의 유사도를 분석하는 베타 다양성 분석 시에도 많은 분석 알고리즘이 있는데,
오늘은 미생물군집의 베타 다양성 분석시 사용할 수 있는 Uniform Manifold Approximation and Projection (UMAP) 에 대해 간단히 소개해보려 합니다..
미생물군집의 샘플 간 유사도를 확인할 수 있는 beta-diversity 분석에는 PCoA (Principal coordinate analysis) 를 사용하여 시각화하는 것이 일반적입니다. UMAP 은 2018 년 ML 관련 논문에서 제시된 기법으로 2024년 기준 인용수 10,000 회를 넘었을 만큼 자주 차용되는 차원축소 기법 중 하나입니다. 최근 single-cell genomics analysis 에서 각광받기 시작한 UMAP 가 상황에 따라 PCoA 를 대체할 수 있는 것으로 보입니다.
PCoA의 단점으로는, 선형 방식으로 한 축에 정사영하여 차원을 축소시키는데, 이 과정에서 아래와 같이 데이터들이 뭉치면서 클러스터가 명확하게 나눠지지 않는다는 것입니다..
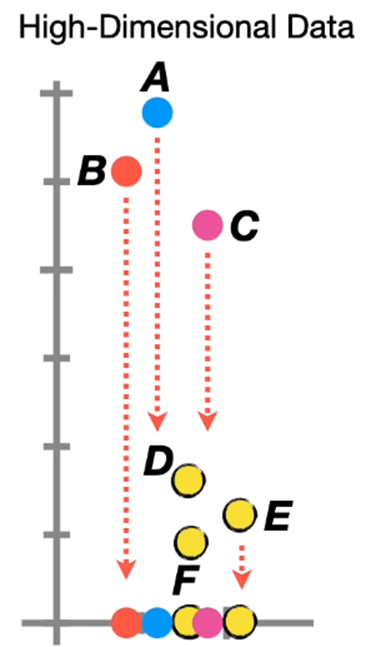
추가로, PCA 등의 기법은 두 개의 주성분이 (PC1, PC2) 대부분의 변동을 설명할 수 있을 때 잘 작동합니다. 그러나 우리의 마이크로바이옴 데이터는 엄청나게 많은 ASVs 들을 feature 로 사용하기 때문에 PCA 등이 잘 작동하지 않을 수 있습니다.
그에 반해, 비선형적 차원 축소 작업을 수행하는 UMAP 은 상대적으로 복잡한 데이터도 잘 처리하며 그 처리속도도 빨라 현재 많이 사용되는 차원축소 알고리즘입니다. UMAP 의 작동 원리에 대해서는 아래 StatQuest 에서 잘 설명해주고 있어 참고하시면 될 것 같습니다.
위 StatQuest 에서 강조하는 것은 number of high-dimensional neighbors 라는 파라미터입니다. 해당 파라미터는 '자신을 포함하여 몇 개의 샘플을 클러스터에 포함되도록 설정할 것인지를 결정' 하며 이는 15 가 기본값이며 사용자의 필요에 따라 조정이 가능합니다. 이를 반영하여 QIIME2 UMAP 에서도 유사한 파라미터를 제공함으로써 필요에 따라 조절할 수 있게 해 줍니다..
UMAP 은 tradeoff 의 특성을 가지고 있는 것이 특징인데, 파라미터를 조정하여 클러스터 안의 샘플 간의 밀도를 강조할 수도 있고 전역적인 거리를 유지하여 클러스터 간의 거리를 보존할 수도 있습니다.
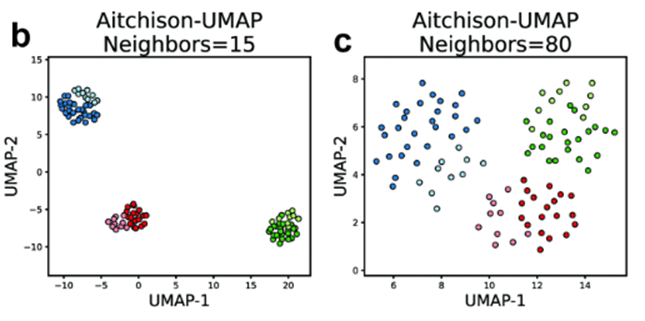
영상에서와 같이 n_neighbors 파라미터에 따라 UMAP 은 한 클러스터에 있는 샘플끼리는 더욱 가깝게,
다른 클러스터에 있는 샘플끼리는 더 멀게 거리를 조정하는 알고리즘을 갖고 있기 때문에 아래 사진과 같이 n_neighbors = 15 인 경우에는 클러스터에 속하는 샘플끼리 더욱 가까워지지만,
n_neigbors = 80 인 경우에는 값이 높아 다른 클러스터에 있는 샘플과도 가까워지도록 알고리즘이 작동하여 클러스터의 구분이 어려워지게 됩니다.
아래의 조금 더 복잡한 데이터셋을 대상으로 PCoA, UMAP 수행한 결과를 보면 확실히 UMAP 의 분류 성능이 더 좋은 것을 확인할 수 있습니다. 개인적인 생각으로는 시간이 갈수록 더욱 활발하게 사용될 알고리즘이 될 것 같습니다.

# reference
1) McInnes, Leland, John Healy, and James Melville. "Umap: Uniform manifold approximation and projection for dimension reduction." arXiv preprint arXiv:1802.03426 (2018).
2) Armstrong, George, et al. "Uniform manifold approximation and projection (UMAP) reveals composite patterns and resolves visualization artifacts in microbiome data." MSystems 6.5 (2021): 10-1128.
'bioinfo' 카테고리의 다른 글
| PICRUST2; Custom database 사용하기 - 1 (중단) (0) | 2024.07.28 |
|---|---|
| FDR correction in LEfSe (0) | 2024.05.12 |
| ECTyper - E. coli serotype identification (1) | 2024.04.27 |
| 인코렌탈 체험, 마이크로바이옴 데이터 간단 분석 (CLC Genomics Workbench Premium , incoRENTAL) (1) | 2024.04.22 |
| DNA Methylation 탐지를 위한 시퀀싱 기법 (0) | 2024.04.07 |